
We are confident that readers will be familiar with the ‘genetic code’ — the concept that the sequence of four bases (A, C, G and T) in DNA is a blueprint for the organism. That is, the machinery of cells can ‘read’ this code, converting sections into proteins — strings of amino acids. Proteins are, as Max Perutz memorably noted, the machines of the cell — in other words they do all the work required for cells, and hence organisms, to function. Our broad theme is, of course, cancer and it is now well-established that chemical changes in DNA — mutations — may alter specific proteins so that they act as ‘drivers’, making cells reproduce (proliferate) abnormally.
However, over the last 80 years it has emerged that cells have a way of modulating gene activity without changing the DNA sequence. The embryologist Conrad Waddington introduced the term ‘epigenetics’ in 1942 and it’s come to mean the study of stable phenotypic changes (known as marks) that do not involve alterations in DNA sequence. In Greek ‘epi’means on or above so that ‘epigenetic’ refers to a kind of fine tuning of gene expression over and above the DNA code. In short epigenetic changes are modifications to DNA that regulate whether genes are turned on or off. There are two types of epigenetic modifications – DNA methylation (direct chemical modification of DNA) and histone modifications (scheme below).

Activation of gene transcription. Top: recognition of DNA sites (nucleotide sequences) near the gene by one or more specific regulatory proteins (transcription factors: green arrows). Blue balls: nucleosomes — segments of DNA wound around eight histone proteins resembling thread wrapped around a spool. Stars: histone modification. The latter permits the chromatin to accommodate RNA polymerase to carry out transcription (green arrow).
In mammals DNA methylation can occur at cytosines anywhere in the genome but more than 98% of DNA methylation occurs in CpG dinucleotides. CpG sites (also called CG sites) are regions of DNA where a cytosine nucleotide is followed by a guanine nucleotide in the linear sequence of bases along its 5′ → 3′ direction. CpG sites occur with high frequency in genomic regions called CpG islands (CG islands). Most CpG dinucleotides are methylated, with the exception of those within CpG islands which are usually unmethylated. We noted the interest in mapping methylation profiles in Cancer GPS? and saw how epigenetic changes affect DNA in Sticky Cancer Genes.
However, hitherto screens of human DNA methylation have covered only a small fraction of the 30 million CpG methylation sites in the human genome and have been limited to cells grown in vitro. Netanel Loyfer and colleagues from The Hebrew University of Jerusalem and other institutes have filled this gap in no uncertain fashion by producing a comprehensive atlas of the human methylome, together with cell type-specific markers and computational tools for the analysis of mixed samples.

Whole-genome DNA methylation atlas of 205 healthy human cell types. From Loyfer et al., 2023.
To acquire this human methylome atlas they used deep whole-genome bisulfite sequencing applied to 39 cell types from 205 healthy tissue samples. As a measure of the precision of their method they showed that replicates of the same cell type were more than 99.5% identical. To analyse the results they divided the genome into almost three million ‘methylation blocks’ (covering at least 3 adjacent CpG sites) that are differentially methylated between cell types. The underlying idea is that functional DNA methylation occurs over regions — methylation blocks — rather than at isolated sites.
Key findings were that methylation patterns were retained during development (ontogeny), i.e. from the time of fertilization of the egg to the adult, and that unmethylated regions in in a cell often occur in transcriptional enhancers and contain DNA binding sites for tissue-specific transcriptional regulators. The latter is consistent with the idea that, broadly speaking, methylation can change the activity of a DNA segment without changing the sequence and, when located in a gene promoter, DNA methylation typically acts to repress gene transcription.
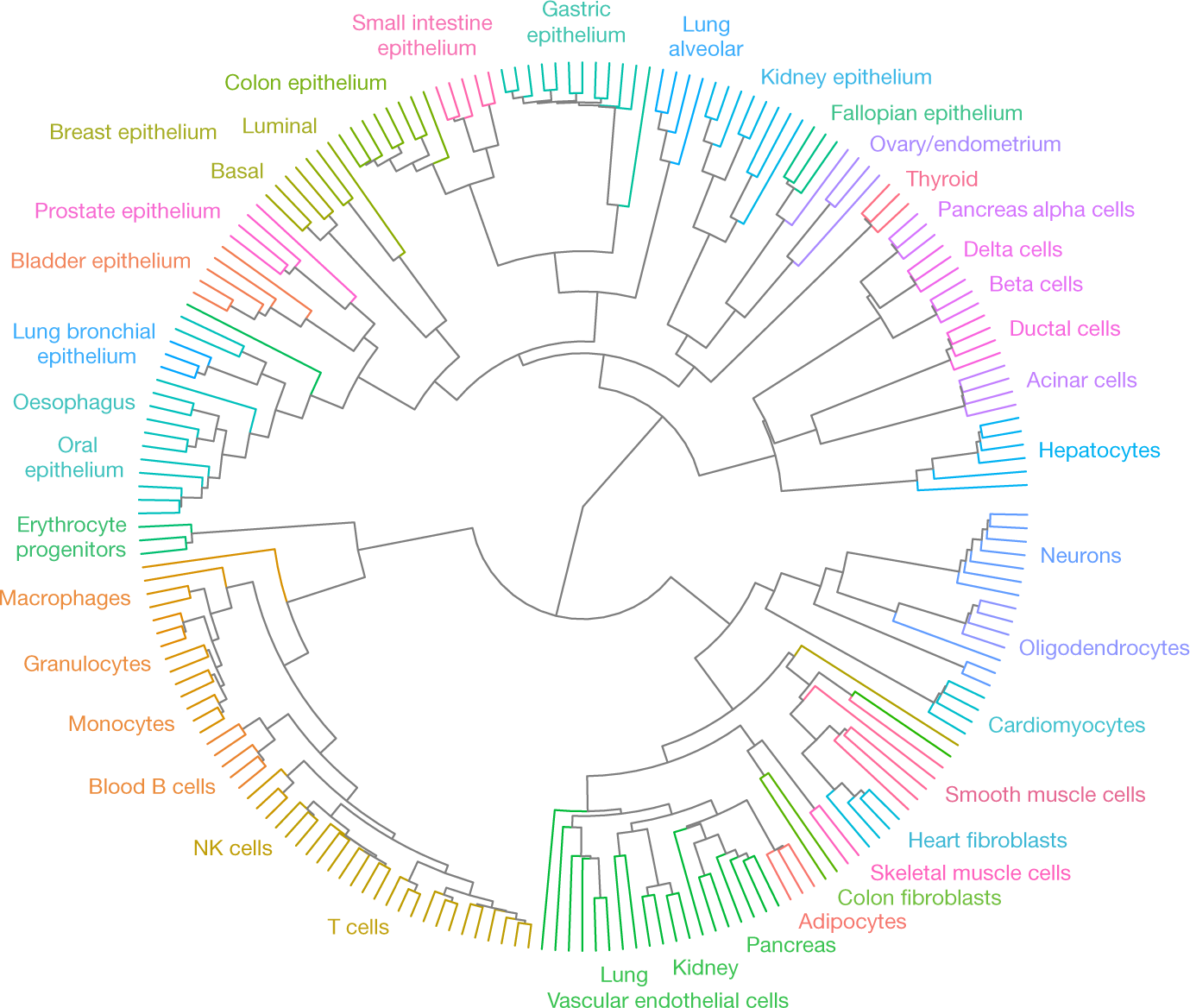
DNA methylation reflects the developmental lineage of healthy human cell types indicated by edge colours. From Loyfer et al., 2023.
The usefulness of this analysis is that the top 25 differentially unmethylated blocks for each cell type yields an atlas of 953 cell type-specific methylation markers that has enormous potential for analysis of composite tissue samples and cell-free DNA.
A further outcome was that cell type-specific unmethylated regions have high levels of DNA accessibility and are enriched for histone marks indicative of active promoters (H3K27ac: an epigenetic modification of histone H3 indicating acetylation of the lysine residue at N-terminal position 27) and enhancers (H3K4me1: mono-methylation at the 4th lysine residue of the histone H3 protein, often associated with gene enhancers).
To confirm the assumption that the differentially unmethylated regions represent gene enhancers they looked for nearby genes with increased expression when the cell type-specific marker is unmethylated. This revealed, e.g., pancreatic islet markers for insulin and glucagon genes (pancreatic islets or islets of Langerhans are the regions of the pancreas that contain endocrine — hormone-producing — cells).
All told the study is a remarkable tour-de-force that has produced a really valuable catalogue of cell type-specific putative promoter and enhancer regions.
Reference
Loyfer, N., Magenheim, J., Peretz, A. et al. A DNA methylation atlas of normal human cell types. Nature 613, 355–364 (2023).
