
These days the notion that cancer cells have picked up genetic alterations (i.e. in their DNA) and that these change the expression of lots of genes is fairly familiar. But what does ‘gene expression’ really mean and how is it controlled? Well, gene expression is the generation of an RNA molecule that carries the same coding information that is enshrined in the sequence of bases in the DNA of that gene — the process of making the RNA is called transcription. The code carried by most RNAs (messenger RNAs — mRNAs) can subsequently be ‘translated’ into the corresponding sequence of amino acids in a protein. And proteins, of course, do all the work to make cells function.
Self-control
And control? There are two levels: 1: the transcription step is regulated to limit the amount of mRNA made from a particular gene. 2: the translation of mRNA into proteins is regulated by post-transcriptional events.
It’s Step 1 that concerns us today in which proteins bind to specific regulatory sequences of DNA in a gene to modulate the activity of the enzyme that makes mRNA molecules from the DNA template. These proteins are generally called transcription factors.
So to be clear: transcription factors are sequence-specific DNA-binding proteins that control the rate of transcription of genetic information from DNA to mRNA — carried out by the enzyme RNA polymerase II. They regulate — turn on or off — genes so that they are expressed in the right cells at the right time. Groups of transcription factors act in a coordinated way to control all cellular processes (growth, division, etc.) and humans have about 1500 of them.
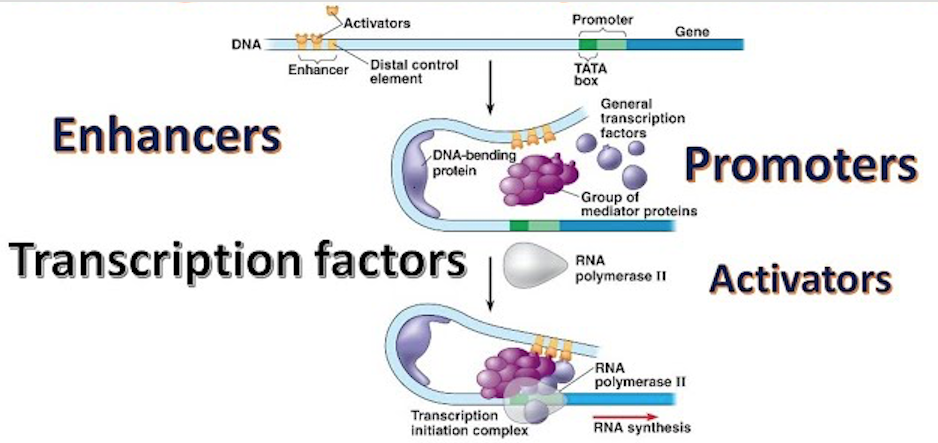
Regulation of gene expression. This scheme shows the concept of regulatory regions within DNA (enhancers and promoters) coming together to control whether a gene is ‘on’ or ‘off’ as a result of transcription factors and mediator proteins binding to specific sequences. When a gene is turned on RNA is synthesised by the action of RNA polymerase II. A TATA box is a sequence of DNA found in the core promoter region of genes. Other proteins (coactivators, chromatin remodelers, histone acetyltransferases, histone deacetylases, kinases and methylases) also regulate gene expression but as these lack DNA-binding domains they are not transcription factors.
Transcription factor of the day
It’s handy to be clear about these basics before we turn to an exciting paper by Sai Gourisankar, Gerald Crabtree and colleagues from Stanford University and the MD Anderson Cancer Center, Houston entitled “Rewiring Cancer Drivers”. They focussed on a transcription factor called B-cell lymphoma-6 (BCL6) that plays an important role in the formation of lymphoid tissues and the production of antibodies. BCL6 normally acts as a repressor — it turns genes off. However, as you’ll guess from its name, BCL6 commonly undergoes mutations in various lymphomas, specifically forms of B-cell non-Hodgkin lymphoma.
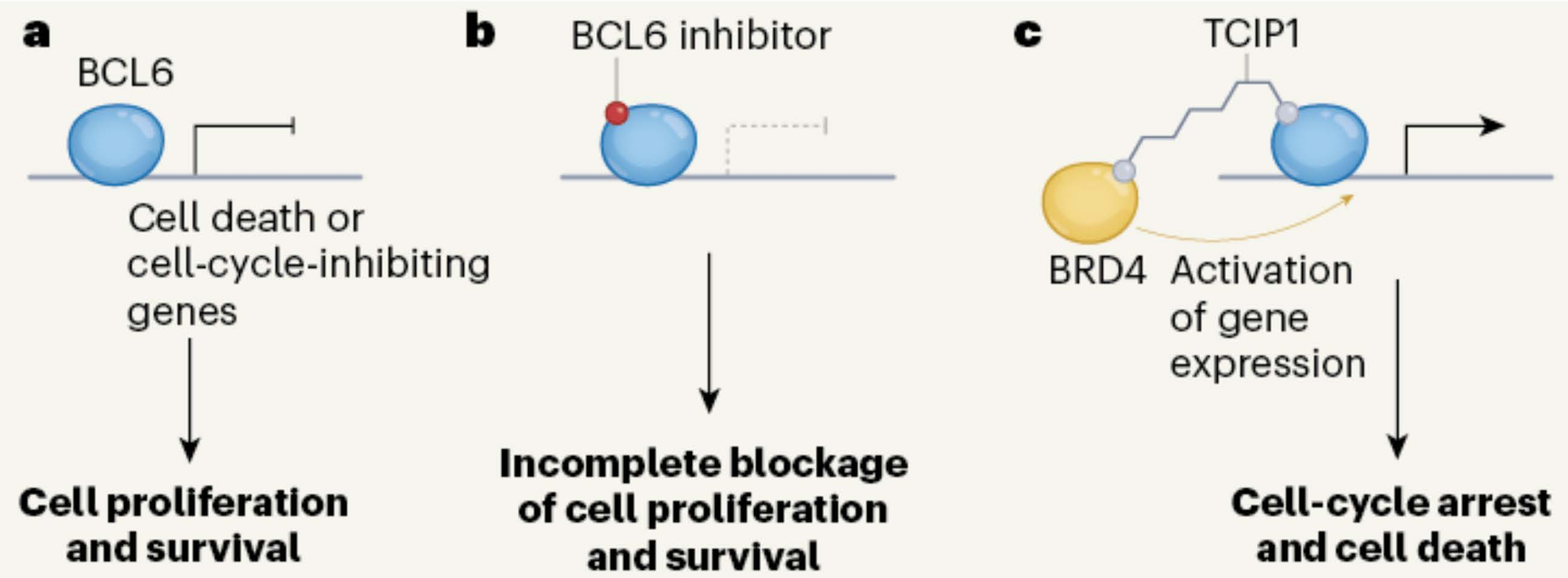
Engineering a transcription factor. a. BCL6 is a transcription factor that, in B cells, normally turns off genes encoding cell cycle inhibitors, thereby allowing some cancer cells to proliferate. b. Small molecule inhibitors of BCL6 have shown some activity as anti-cancer agents. c. Chemical coupling of TCIP1to BCL6 recruits BRD4, a transcription activator. This gives strong activation of genes normally turned off by BCL6 and kills cancer cells expressing BCL6. From Phelan and Staudt, 2023.
Why it’s exciting
Scheme c above shows Gourisankar & Co’s clever idea. They coupled BCL6 to a small molecule called TCIP1 (if you’re really interested this stands for transcriptional/epigenetic chemical inducer of proximity). TCIP1 binds to the protein BRD4 (bromodomain-containing protein 4) — a transcriptional and epigenetic regulator that plays a critical role in embryogenesis and cancer development. This juxtaposes BRD4 and BCL6, the upshot being that BRD4 potently activates the expression of genes normally silenced by BCL6.
Great chemistry — so what?
TCIP1 is an extremely potent killer of diffuse large B cell lymphoma cells that express BCL6. Thus the approach of using TCIP1 offers a new therapeutic strategy for these cancers. However, it may also be effective against other cancer types and there is good reason for optimism because there was a bonus in this paper in that TCIP1 also downregulates MYC — a master regulator that is abnormally expressed in many cancers. It’s not clear how this works as BRD4 is generally considered to drive cancers by activating MYC — so the mechanism is indirect but nontheless welcome for that!
And the down-side?
The flip side is that BCL6 suppresses inflammation in immune cells (knocking out BCL6 kills mice via an inflammatory reaction) which might be a problem for a treatment that targets this transcription factor. However, the authors looked hard in both normal mice and in human cells (fibroblasts) for evidence of tissue damage and found none, even though there were large changes in gene expression in spleen and lymphocyte cells.
Clearly there is much to learn about this approach of using what is in effect a double-headed molecule as an anti-cancer agent. The name of the Roman god of beginnings and endings has already been nicked by molecular biologists for the JAK kinases (their two faces link cytokine receptors to other proteins). Let us therefore avoid giving a trendy name to this new approach using TCIP1 but perhaps just nod to Janus in the hope that it will become a useful anti-cancer strategy.
References
Phelan JD, Staudt LM. Double-headed molecule activates cell-death pathways in cancer cells. Nature. 2023 Aug;620(7973):285-286. doi: 10.1038/d41586-023-02213-4. PMID: 37495782.
Gourisankar S, Krokhotin A, Ji W, Liu X, Chang CY, Kim SH, Li Z, Wenderski W, Simanauskaite JM, Yang H, Vogel H, Zhang T, Green MR, Gray NS, Crabtree GR. Rewiring cancer drivers to activate apoptosis. Nature. 2023 Aug;620(7973):417-425. doi: 10.1038/s41586-023-06348-2. Epub 2023 Jul 26. Erratum in: Nature. 2023 Sep;621(7977):E27. PMID: 37495688.



